
The Drug Development Playbook - Part 6
Author: Aryan Kenia
Life Sciences Analyst
This is Part 6 of The Drug Development Playbook. In Parts 1 to 5 we mapped the pipeline, explained target choice, showed how chemists convert hits to candidates, walked the candidate through preclinical gates, and described clinical design. Now you have pivotal data. This article explains how to turn that data and your manufacturing story into an approval. I focus on practical filing gates, dossier strategy, review risk management, and commercial alignment so you can make the filing decision like a project lead with both science and business in mind.
Get the companion PDF – The Consultant’s Companion: Actionable Frameworks from The Drug Development Playbook
This short guide collects the practical checklists and decision frameworks I refer to across the series. Use it in meetings, as a quick audit, or to run go/no-go gates when you don’t want to guess.
Key Summary
- A submission is two things at once: a technically perfect eCTD and a sharp Module 2 narrative that argues benefit versus risk.
- Expect FDA clocks of about 6 months for priority review and 10 months for standard review; EMA uses a 210 active day scientific review plus the Commission decision window.
- The hardest, most value-at-risk work lies in Module 3 CMC comparability, Module 2 storytelling, and inspection readiness. Get those right before you file.
Acronyms used in this article
- CTD: Common Technical Document
- eCTD: Electronic Common Technical Document
- NDA: New Drug Application
- BLA: Biologics License Application
- MAA: Marketing Authorisation Application
- PDUFA: Prescription Drug User Fee Act (FDA review timelines)
- REMS: Risk Evaluation and Mitigation Strategy (US)
- RMP: Risk Management Plan (EU)
- CHMP: Committee for Medicinal Products for Human Use (EMA)
- PRAC: Pharmacovigilance Risk Assessment Committee (EMA)
- PRIME: PRIority MEdicines (EMA)
- GMP: Good Manufacturing Practice
- GLP: Good Laboratory Practice
- PV: Pharmacovigilance
- HTA: Health Technology Assessment
- PMR/PMC: Post-Marketing Requirement / Post-Marketing Commitment
- PI: Prescribing Information (US)
- SmPC: Summary of Product Characteristics (EU)
A short hypothetical case study
A small biotech ran two positive pivotal trials in a chronic inflammatory disease. Their clinical data sits well, but their API route changed between Phase II and Phase III to improve yield. The regulator flags product comparability in the first round of questions. The sponsor wastes six months doing bridging analytics and a GLP tox bridging study while a competitor files. That delay erodes market exclusivity and valuation. The moral is simple. Filing without a synchronized CMC narrative and inspection readiness risks the whole program, not just the clock.
The dossier versus the narrative
Regulators don’t approve a single study or a single batch. They approve an application that tells a coherent story: what the product does, why it helps patients, and why manufacturing and controls ensure safety and quality. The CTD is the agreed format for that story. The electronic CTD must validate technically, but reviewers read Module 2 first and use it to decide where to dive deeper. So you must produce two deliverables in parallel:
- a compliant eCTD that passes technical validation and fileability checks (XML structure, no broken links), and
- a tight Module 2 narrative that answers the regulator’s likely questions about benefit, risk, and quality.
If you focus on eCTD packaging at the expense of the narrative you will invite questions; if you perfect the narrative but leave CMC or inspection readiness weak you will invite a CRL. Balance both.
Key documents that reviewers use to form a first impression: Module 2 Clinical Overview and Integrated Summaries of Efficacy and Safety. These are your executive briefings. Make them read like answers to the regulator’s top concerns.
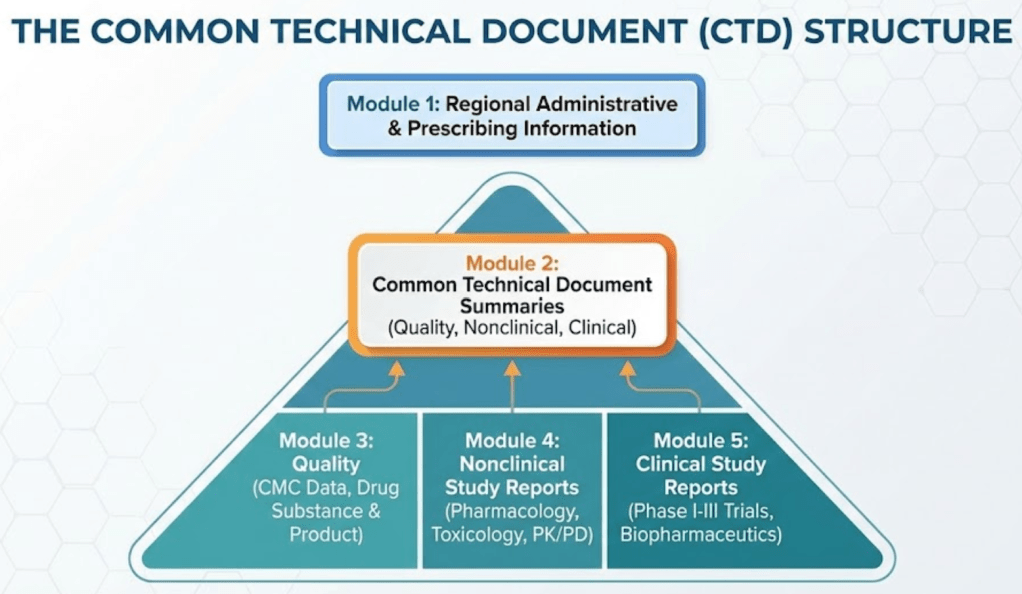
The CTD in plain English
The CTD divides the submission into five modules:
- Module 1: region specific administrative documents and proposed label.
- Module 2: summaries and the narrative core — Quality Overall Summary, Nonclinical Overview, Clinical Overview, ISE and ISS.
- Module 3: full CMC data — manufacturing, controls, stability, impurity profiles. This is the place that often triggers review holds.
- Module 4: nonclinical study reports. Reviewers target pivotal GLP studies and the data used to justify starting doses.
- Module 5: clinical study reports, including pivotal Phase III trials and integrated analyses.
Think of Module 2 as the story and Modules 3 to 5 as the evidence vaults. Write Module 2 to direct reviewers to the evidence they need and to pre-empt the questions they will ask.
Authoritative CTD guidance comes from ICH and regional agencies. Use ICH M4 as your formatting and content blueprint.
Regulatory pathways and what they mean for filing strategy
Short timeline facts that change planning:
- FDA standard review aims for action in around 10 months after filing; priority review shortens that goal to about 6 months. These clocks still allow for information requests and clock stops.
- EMA centralized review follows a 210 active day scientific evaluation with clock stops; the Commission decision adds a further administrative window.
Choose the pathway that matches your value and data strength. Accelerated or conditional approvals can speed access, but they usually add more post-approval obligations and conditional commitments. If you plan to request PRIME or Breakthrough designation, map how that changes interactions and expectations for post-approval confirmatory evidence.
The review process step by step
Fileability check and day 60 filing decision. If the agency accepts the file they set the review clock and assign discipline leads. During review multidisciplinary teams examine Module 2 and then jump into Modules 3 to 5 for the evidence. Expect iterative information requests, mid-cycle interactions, and possible advisory committee preparation if the benefit-risk profile raises public or scientific debate.
Manufacturing inspections and GCP audits can happen concurrently. Regulators may inspect one or more manufacturing sites and pivotal clinical sites. If inspections find deficiencies that impact product quality or data integrity, agencies will raise those as regulatory holds or CRLs.
Practical implication: plan for inspection readiness weeks before filing and have CAPA plans that you can show regulators quickly.
Labeling and indication strategy
Approval is not simply yes or no. The label becomes the legal description of your product’s use and it drives commercial value. During pre-submission meetings draft realistic PI and SmPC texts and map how each proposed claim affects reimbursement and market size. Ask these questions early:
- Do you want a biomarker-defined label or an all-comers label?
- Will you accept narrower wording to speed approval?
- Does your label support the HTA evidence needed for pricing?
Label negotiation often defines who will use the drug, not just whether it will be allowed. Test narrow and broad label scenarios in rNPV models so you know what to accept and what to fight for.
Advisory committees
Advisory committees present high-visibility risk and opportunity. Agencies convene them when data raise hard questions or when public interest is high. Prepare like you would for a court case:
- build a balanced briefing book that highlights both strengths and limitations,
- run multiple mock panels, and
- train spokespeople to answer blunt, uncomfortable questions succinctly.
A failed advisory meeting can mean long delays or a narrowed label. Treat the meeting as a controlled crisis that you must win with clarity and humility.
Manufacturing, comparability, and CMC risk
Most late-stage delays come from CMC issues. Regulators expect to see that your commercial process makes product that matches critical quality attributes from clinical batches. If you changed the route, site, or formulation, prepare a comparability package now: analytical panels, impurity comparisons, and a plan for any nonclinical bridging that regulators may request.
Validate key analytics, run stability studies sufficient for initial shelf life, and prepare PPQ plans. For biologics expect inspectors to probe process control, viral safety, and product heterogeneity closely.
If you cannot complete some commercial validation before filing, document why, present timelines, and include risk mitigations to maintain credibility.
Risk management: REMS, RMP, and market access implications
Regulators use REMS in the US and RMP in the EU to manage residual risks. REMS with Elements To Assure Safe Use can restrict access and increase distribution complexity. That restriction has commercial consequences.
Model the impact of a REMS or RMP on uptake and pricing. If your safety profile risks a REMS, propose phased approaches and evidence collection plans that reduce burden while protecting patients. The FDA provides guidance documents on REMS content and modification that you should read early.
Data integrity, inspections, and auditing
Inspectors will look at ALCOA plus principles for clinical and manufacturing records. Validate electronic systems, preserve audit trails, and resolve data queries long before filing. A late discovery of retrospective entries or poor audit trails can trigger a wide data integrity investigation and delay approval.
Simulate inspections with independent auditors, and track CAPAs to closure. Keep a clean, defensible trail for your Integrated Summary of Safety and Clinical Study Reports.
HTA and payer alignment
Regulators judge benefit and risk. Payers judge value and affordability. Align clinical endpoints, comparators, and health economic inputs with HTA expectations. If your pivotal trials used a surrogate endpoint that regulators accept, check whether payers will call for additional outcomes. Use parallel scientific advice where available and prepare early payer dossiers for key markets.
Post-approval commitments and lifecycle planning
Approval starts lifecycle management. Accelerated approvals almost always require confirmatory trials. REMS and RMPs require ongoing surveillance and resources. Use ICH Q12 to design CMC change paths that reduce future regulatory friction. Plan for label expansions and build registries or real-world evidence collection into launch budgets.
Filing gates you must meet before you press submit
Make a heatmap with the following gates and owners. If any gate sits red, do not file until you have a credible remediation plan and timeline.
1. Clinical gate (CMO)
- primary endpoints met per SAP, ISE and ISS drafted and consistent.
2. Statistical gate (Head Biostatistics)
- all planned analyses complete, sensitivity analyses done, estimands defined.
3. CMC gate (Head Technical Ops)
- commercial process validated or PPQ plan approved and comparability data ready.
4. Manufacturing gate (Quality)
- key sites audit-ready, CAPAs closed or on track.
5. Regulatory gate (Head Regulatory Affairs)
- pre-NDA/MAA meetings complete, eCTD validation runs clean.
6. Commercial/HTA gate (Head Market Access)
- draft label supports payer dialogue, evidence gaps mapped.
7. eCTD/Operations gate (Reg Ops)
- XML validation passes, no broken links, file structure final.
Common late-stage failure modes and how consultants prevent them
- CMC comparability failures: fix by early pilot batches and analytical comparability plans.
- Inconsistent pivotal results: pre-submit pooled and sensitivity analyses and reconcile differences with regulators.
- Inspection findings: run mock inspections and audit CRO/CDMO partners well before filing.
- Label negotiation losses: prepare narrow and broad label scenarios and model value consequences.
A consultant’s job is to force these hard conversations early and to present regulators with clear remediation paths rather than surprises.
Next in the series
Part 7, the final part of this series, talks about what comes after approval. Post‑approval is less about molecules and more about systems, signals, and evidence strategy.
Parts of this series
- Part 1 – From Idea to Patient: How Drug Development Actually Works
- Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- Part 3 – Hit Discovery and Lead Optimization – how teams turn a validated target into an actual drug candidate
- Part 4 – Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people
- Part 5 – Clinical Development: Phases I to III
- Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy
- Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle
References
- ICH. The Common Technical Document (CTD). ICH M4 guidance.
- FDA. Priority Review, Fast Track, Breakthrough Therapy, Accelerated Approval guidance and PDUFA review timelines.
- EMA. Centralised procedure timelines and PRIME scheme guidance.
- FDA. Risk Evaluation and Mitigation Strategies (REMS) guidance and format/content documents.
- EMA. Guidance on preparing for PRIME scheme access and submission readiness.

Leave a reply to After Approval: Running Safety, Real World Evidence, and the Product Lifecycle – Aryan Kenia Cancel reply