
The Drug Development Playbook - Part 4
Author: Aryan Kenia
Life Sciences Analyst
This is Part 4 of The Drug Development Playbook. Part 1 mapped the full journey, Part 2 picked the biology, and Part 3 made the chemistry. Now we build the nonclinical and CMC evidence that lets regulators say yes to first-in-human trials.
Get the companion PDF – The Consultant’s Companion: Actionable Frameworks from The Drug Development Playbook
This short guide collects the practical checklists and decision frameworks I refer to across the series. Use it in meetings, as a quick audit, or to run go/no-go gates when you don’t want to guess.
Key Summary
- Preclinical is the first hard regulatory gate. Get this wrong and regulators will stop your trial, or you’ll spend months and millions repeating studies.
- Regulators want four things: a safety margin, PK/PD that ties your biology to dose, a CMC story that ensures clinical material is consistent, and a translational rationale for the FIH plan.
Acronyms used in this article
IND: Investigational New Drug application
CTA: Clinical Trial Application
FIH: First-in-Human
MRSD: Maximum Recommended Starting Dose
MABEL: Minimum Anticipated Biological Effect Level
NOAEL: No Observed Adverse Effect Level
HED: Human Equivalent Dose
HNSTD: Highest Non-Severely Toxic Dose
STD10: Severe Toxic Dose in 10% of animals
GLP: Good Laboratory Practice
GMP: Good Manufacturing Practice
CMC: Chemistry, Manufacturing, and Controls
API: Active Pharmaceutical Ingredient
DMPK: Drug Metabolism and Pharmacokinetics
TK: Toxicokinetics
PK/PD: Pharmacokinetics / Pharmacodynamics
ADME: Absorption, Distribution, Metabolism, Excretion
DDI: Drug-Drug Interaction
DART: Developmental and Reproductive Toxicology
ICH: International Council for Harmonisation
EMA: European Medicines Agency
FDA: Food and Drug Administration
OECD: Organisation for Economic Co-operation and Development
LC-MS/MS: Liquid Chromatography with Tandem Mass Spectrometry
LLOQ: Lower Limit of Quantitation
Caco-2: Caco-2 permeability assay
PAMPA: Parallel Artificial Membrane Permeability Assay
MDCK: Madin-Darby Canine Kidney cell permeability assay
HTS: High-Throughput Screening
FBLD/FBDD: Fragment-Based Lead Discovery
SBDD: Structure-Based Drug Design
PoC: Proof of Concept
PoM: Proof of Mechanism
TPP: Target Product Profile
rNPV: Risk-Adjusted Net Present Value
LOA: Likelihood of Approval
NME: New Molecular Entity
CRO: Contract Research Organization
CDMO: Contract Development and Manufacturing Organization
A short scene that matters
A program readied for clinical dosing until the CRO’s GLP histopathology report showed a different impurity profile in the tox material versus the planned clinical material. Regulators flagged comparability issues. The company had to run bridging tox with the clinical GMP batch. The hold dragged the program by nine months and pushed them out of an important partnership window.
When you get to preclinical, the small stuff becomes the big stuff. Most delays come from gaps in sequencing, documentation, or CMC, not from mysterious new toxicities.
Why preclinical is the gating stage
Preclinical development turns a development candidate into an evidence package suitable for an IND in the US or a CTA in Europe and elsewhere. Regulators look at that package and ask one question: given the planned first-in-human study, do the benefits and knowledge justify the risks? If your package fails to answer that question, you get a clinical hold or an extra study requirement. Preclinical is not a box-checking exercise. You must show scientifically justified safety margins, PK/PD, and well-controlled material.
What regulators expect to see
Regulators review your dossier as an integrated story that ties molecule to clinic. The four pillars are:
- Nonclinical pharmacology – primary pharmacology and relevant off-target profiling.
- Toxicology and safety pharmacology – GLP pivotal repeat-dose studies plus core safety pharmacology per ICH S7A/S7B.
- DMPK/ADME and toxicokinetics – cross-species PK, metabolite ID, exposure multiples.
- CMC – a controlled route of manufacture, specifications, and stability that make the clinical batch defensible.
Regulators always read those four elements against the FIH plan: route, duration, dose escalation, and subject population.
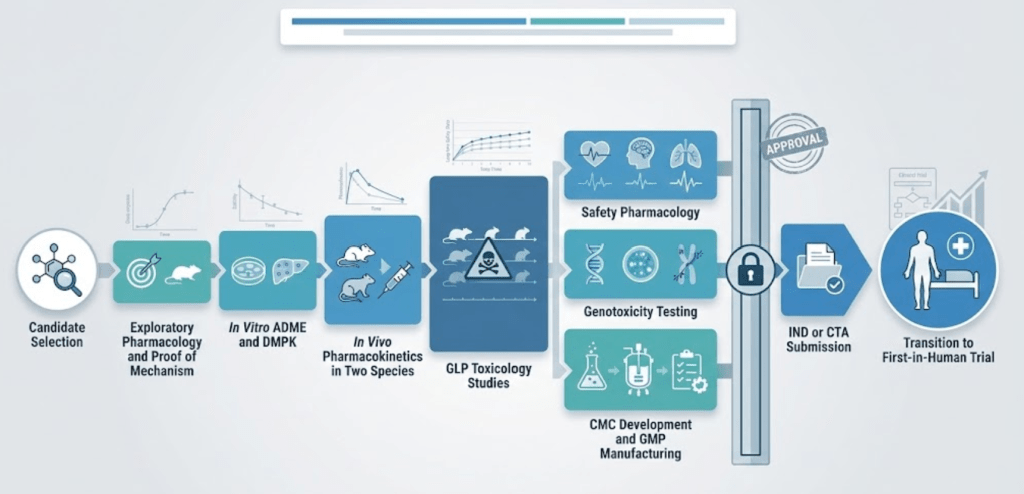
Typical preclinical flow and practical timelines
A standard small-molecule IND package usually follows this sequence:
- Exploratory pharmacology and proof-of-mechanism studies.
- In vitro ADME and quick DMPK screens.
- In vivo PK in two species.
- GLP repeat-dose toxicology in two species, with TK.
- Safety pharmacology core battery and genotoxicity.
- Stability and initial GMP/CMC work for clinical material.
Expect focused IND-enabling work to take on the order of months, not weeks. Industry commonly plans 9-18 months for the formal IND-enabling nonclinical work and CMC scale-up, but timelines vary with modality and scope. Poor sequencing causes most delays: e.g., CMC lagging behind tox or biomarkers not ready when the clinical protocol needs them.
Pharmacology and translational PD: make the FIH hypothesis tight
Primary pharmacology must show your molecule engages the target and produces the expected downstream effect in human-relevant systems. That means binding and functional assays in vitro, plus in vivo PoM or PoC in models that you can justify as translational.
Secondary pharmacology covers off-target screening. Use receptor and ion-channel panels to catch liabilities that would require specific clinical monitoring, like neuro or cardiac effects.
Biomarkers and PK/PD models are your currency with regulators. If you can show target engagement and a dose → exposure → PD → effect chain in animals, you can propose a reasoned MABEL or MRSD for FIH. Invest in translational biomarkers early. Regulators expect the rationale to be quantitative, not narrative.
DMPK and bioanalysis: exposure equals safety and efficacy
Before you dose people, understand how the molecule behaves:
- In vitro ADME gives metabolic stability and DDI flags.
- In vivo PK in at least two species gives clearance, half-life, and bioavailability.
- Toxicokinetics (TK) in GLP tox studies lets you link toxic findings to exposure and calculate margins.
- Bioanalytical methods must be validated to quantify parent and major metabolites at the expected levels.
If you can’t show exposure in animals that models the intended human exposure, pause. Potency alone won’t translate if you lack drug in the target tissue.
GLP toxicology and safety pharmacology
Core elements for small molecules:
- Repeat-dose tox in two species with TK. Choose species for pharmacologic relevance and metabolic comparability.
- Safety pharmacology per ICH S7A: cardiovascular, CNS, and respiratory endpoints. For QT risk, align with S7B approaches.
- Genotoxicity: Ames plus in vitro mammalian cell tests; follow with in vivo tests if needed.
- Reproductive tox is staged and aligned to the clinical plan; not all DART tests are needed for an FIH if women of childbearing potential are excluded, but you must plan for eventual inclusion.
For biologics, ICH S6(R1) lets you focus on the single pharmacologically relevant species and demands immunogenicity and target-mediated safety assessments.
GLP, non-GLP, NAMs and smart sequencing
Pivotal studies supporting the IND must be GLP or GLP-equivalent. Exploratory studies can be non-GLP, and regulators accept well-justified non-animal methods where validated. Smart teams front-load non-GLP or NAMs to de-risk the GLP program and reduce animal use, but they deliver the GLP package on time. OECD GLP principles remain the cornerstone for data quality.
CMC basics that regulators read as safety data
Regulators treat CMC like a safety argument for early trials: can you make clinical material consistently so that subjects are not exposed to unexpected impurities or variants?
Key items for IND/CTA readiness:
- Drug substance identity, assay, and impurity profile.
- Drug product formulation, container-closure, and stability data that support the intended shelf life for the trial.
- Comparative data that justify the use of nonclinical material versus the planned clinical GMP batch; if you see differences, plan bridging studies now.
- Basic GMP manufacture of clinical material and defined release testing.
Don’t discover a different polymorph or a new impurity only when you file. That causes holds and bridging requests.
Dose selection: NOAEL, HED, MRSD, and MABEL
Regulators expect a quantitative starting dose rationale.
- For many small molecules, convert animal NOAEL to a Human Equivalent Dose (HED) and apply a safety factor to derive an MRSD. The FDA provides an accepted algorithm for this.
- For high-risk biologics or novel mechanisms, compute a MABEL using in vitro receptor occupancy, animal PK/PD, and human target properties to select an appropriately low starting dose.
- Oncology uses alternative paradigms (HNSTD, STD10) with higher tolerated risks in many contexts.
Pair dose logic with sentinel cohorts and predefined stopping rules tied to exposure multiples.
Tactical regulatory interactions
Book pre-IND or scientific advice early. Use those meetings to align species choice, tox packages, and your CMC comparability plan. Regulators do not accept surprises; they tolerate justified risk. A well-structured pre-IND package and specific questions shorten reviews and reduce the chance of a clinical hold. Timelines differ across regions, but early engagement always helps.
Operational traps that slow programs
Most program delays come from logistics, not science:
- CRO selection mismatch for species or GLP capability.
- Bioanalytical methods delivered late.
- GMP batches available after GLP tox completes, creating comparability gaps.
- Long-lead items like special animals, radiochemistry, or labeled compounds.
Mitigate these by mapping critical path dependencies in your IND Gantt and holding owners accountable.
Decision gates and kill criteria – be ruthless and rational
Predefine go/no-go gates tied to safety margins, PK, and CMC comparability. Example gates:
- Tox: unacceptable, non-reversible lesions at low multiples or organ effects without monitoring or mitigation.
- PK: inability to reach projected efficacious exposure without hitting tox limits.
- CMC: irreproducible clinical material or unsafe impurity profile.
Clear gates protect portfolio value. Killing early often preserves more upside for the company than a late, expensive failure.
Costs and ballpark timelines
Numbers vary with modality and scope, but industry sources commonly place IND-enabling nonclinical and CMC efforts in the low millions. Expect 9-18 months for focused IND enabling work for a small molecule and plan budgets accordingly. Get firm CRO and CDMO quotes early; vendor variance drives schedule risk.
What to expect next
Part 5 talks about the transition to in-human clinical trials and what happens from phase I to III.
Parts of this series
- Part 1 – From Idea to Patient: How Drug Development Actually Works
- Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- Part 3 – Hit Discovery and Lead Optimization – how teams turn a validated target into an actual drug candidate
- Part 4 – Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people
- Part 5 – Clinical Development: Phases I to III
- Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy
- Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle
References
- ICH M3(R2). Non-clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. International Council for Harmonisation (ICH). Step 5, 2009. PDF.
- ICH S7A. Safety Pharmacology Studies for Human Pharmaceuticals. International Council for Harmonisation (ICH). 2001. PDF.
- ICH S7B. Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation). International Council for Harmonisation (ICH). 2005. PDF.
- ICH S6(R1). Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. International Council for Harmonisation (ICH). 2012 (R1 addendum). PDF.
- ICH Q1A(R2). Stability Testing of New Drug Substances and Products. International Council for Harmonisation (ICH). PDF.
- FDA. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Guidance for Industry. U.S. Food and Drug Administration (FDA), July 2005 (updated pages online).
- EMA. Guideline on Strategies to Identify and Mitigate Risks for First-in-Human and Early Clinical Trials with Investigational Medicinal Products (Revision 1). European Medicines Agency (EMA), 2017. PDF.
- OECD Principles of Good Laboratory Practice (GLP) and Compliance Monitoring. Organisation for Economic Co-operation and Development (OECD). PDF.
- ICH Q2(R1/R2). Validation of Analytical Procedures: Text and Methodology. International Council for Harmonisation (ICH). PDF.
- FDA. Bioanalytical Method Validation Guidance for Industry. U.S. Food and Drug Administration (FDA), May 24, 2018. PDF.
- ICH Q7. Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients (APIs). International Council for Harmonisation (ICH). PDF and WHO/FDA pages.
- FDA. Bioanalytical Method Validation and Related Topics (M10; draft/final materials as applicable). U.S. Food and Drug Administration (FDA). PDF, 2024.
- ICH E14 / S7B Q&As – Integrated considerations for QT assessment. International Council for Harmonisation (ICH). PDF Q&A.
- Gabrielsson J., Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications. 5th ed. Routledge / Apotekarsocieteten, 2016. (Good primer on PK/PD modelling and translation.)
- Review – Allometric scaling and interspecies prediction in drug development. van Valkengoed DW et al. “All You Need to Know About Allometric Scaling.” Integrative review, 2024. PMC.
- Practical CRO/CDMO guidance and IND-enabling planning pages (example industry resources): Charles River IND enabling pages and CRO planning overviews.

Leave a reply to After Approval: Running Safety, Real World Evidence, and the Product Lifecycle – Aryan Kenia Cancel reply