The Drug Development Playbook - Part 3
Author: Aryan Kenia
Life Sciences Analyst
This is Part 3 of The Drug Development Playbook. Part 2 picked the biology worth backing. Now we answer the next question: how do you find molecules that not only hit that biology but can become safe, manufacturable medicines?
Get the companion PDF – The Consultant’s Companion: Actionable Frameworks from The Drug Development Playbook
This short guide collects the practical checklists and decision frameworks I refer to across the series. Use it in meetings, as a quick audit, or to run go/no-go gates when you don’t want to guess.
Key Summary
- Hit discovery finds anything that modulates your target; lead optimization sculpts that chemistry into a candidate that works in people and in a factory.
- Treat discovery as a multi-parameter optimization problem, not a pure potency contest.
- One practical step: build a clear candidate profile up front and force every design cycle to ask whether a change moves you closer to that profile.
A short scene from a lab
I once saw a LinkedIn post where the chemistry team proudly produced a compound with 2 nM potency. The assay looked great. The slides looked great. The CEO asked about oral exposure in rats. The chemists had no PK data yet. Two months later, the same series failed repeatedly in clearance assays. They had optimized potency but ignored exposure. Weeks of work and a good-looking molecule became a sunk cost.
That moment taught me this rule: potency without exposure is a beautiful dead end.
Where “hit discovery and lead optimization” fits in the pipeline
Hit discovery finds chemical matter that reproducibly affects the target, often at micromolar potency and without deep ADME data. Hit-to-lead confirms hits, expands around series, and builds early SAR. Lead optimization then runs intensive medicinal chemistry cycles to balance potency, selectivity, PK, safety, and developability until you have a single development candidate ready for GLP tox and process scouting.
In short, you move from “does anything bind?” to “can we sculpt a molecule that does everything we need in a human body and a factory?”
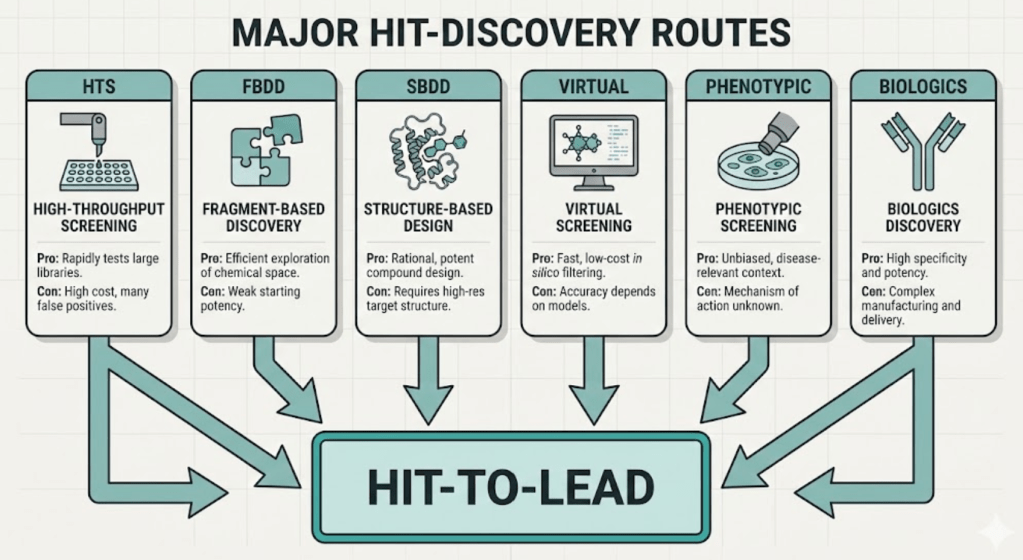
The main discovery routes
High-throughput screening (HTS)
HTS screens large libraries, from tens of thousands up to millions of compounds, in a standardized assay to find initial hits. HTS finds diverse chemotypes and can uncover unexpected starting points. But it also throws up many false positives and compounds that create downstream headaches. Run orthogonal assays and counterscreens early. HTS works well for well-behaved biochemical targets and when you want breadth.
Fragment-based lead discovery (FBLD)
Fragments use tiny, low-molecular-weight pieces that bind weakly but efficiently. You detect binding with sensitive biophysics like NMR or X-ray, then grow, link, or merge fragments into potent leads. FBLD explores chemical space efficiently and often yields clean, high-efficiency leads, especially for hard pockets or protein–protein interfaces. Expect to invest in structural biology and biophysics to make this pay.
Structure-based and virtual approaches
If you have structural data, use it. Co-crystal structures accelerate SAR by showing how modifications change binding. Docking and virtual screens can triage millions of candidates cheaply, especially if you feed back experimental data to refine models. Don’t treat docking scores as gospel; use them to focus synthesis and testing.
Phenotypic and biologic discovery
Phenotypic screens find compounds that change a disease-relevant readout. They can create first-in-class opportunities but require deconvolution to identify the mechanism. For biologics, discovery emphasizes epitope, affinity, and developability rather than classic SAR. Treat all discovery routes as hypothesis-generating, then test those hypotheses against human-relevant biology.
Hit-to-lead
Not every hit becomes a lead. Hit-to-lead focuses on:
- Confirming activity with orthogonal assays.
- Removing assay artifacts and PAINS.
- Building early SAR to see whether potency and selectivity respond to modification.
- Running quick ADME screens (solubility, microsomal stability, permeability) to flag obvious dead ends.
Hit-to-lead is a low-cost, high-information stage. It gives you a sense of whether a chemotype has room to evolve or whether you should kill it and move on.
Lead optimization
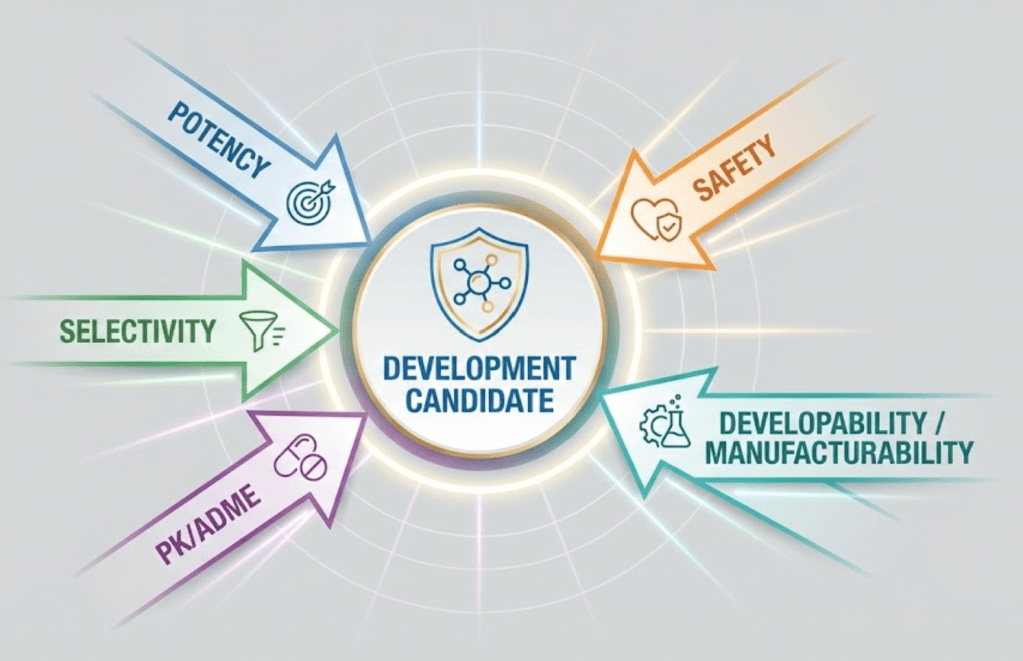
Medicinal chemistry during lead optimization juggles several properties simultaneously:
- Potency and selectivity versus key off-targets.
- Sufficient exposure at the site of action (oral bioavailability, half-life, clearance).
- Safety flags such as hERG liability and CYP interactions.
- Physicochemical properties for formulation and solid-state behavior.
- Synthetic tractability and cost.
Successful teams integrate biology, DMPK, and analytical support with chemistry, and they run tight design–make–test cycles to move several properties together. Don’t optimize potency at the expense of PK or developability. That’s the fastest route to a dead end.
ADME and PK levers you must track
Early DMPK data guide chemistry choices. Key levers include:
- Oral bioavailability: depends on solubility, permeability, first-pass metabolism. Measure early with in vitro (Caco-2, microsomes) and quick in vivo rodent PK.
- Clearance: high clearance kills exposure. Identify metabolic soft spots and block them with bioisosteres or steric hindrance.
- Volume of distribution: lipophilicity and basicity influence tissue partitioning. Aim for the Vd appropriate to your indication.
- First-pass metabolism: if you see heavy gut or hepatic extraction, explore prodrugs, alternate routes, or different chemotypes.
Tie each design decision to a target product profile: what exposure do you need, and can the chemistry plausibly achieve it?
Early safety screens that save time and money
Catch common liabilities early:
- hERG cardiac risk: screen binding or channel activity early, and use ICH S7B principles to interpret risk. Reducing lipophilicity and removing planar basic motifs helps.
- CYP inhibition and induction: profile major CYP isoforms to avoid later drug–drug interaction surprises.
- Off-target panels: run common receptor panels to flag undesirable CNS or cardiovascular interactions.
- Genotoxicity flags and reactive metabolite potential: use in silico alerts and quick in vitro tests to avoid carrying toxicophores.
These screens help you apply rational kill criteria and avoid “zombie” series that consume capital.
Developability and manufacturability matter early
Don’t treat solid-state, formulation, or synthetic route as afterthoughts. Consider:
- Solid form and polymorphism risks.
- Salt and co-crystal options to improve solubility.
- Synthetic route length, chiral complexity, and supply chain for starting materials.
- Cost of goods for the intended market and dosing regimen.
A brilliant molecule that needs exotic chemistry or yields a nightmare solid form can destroy NPV. Build process chemists into candidate selection early.
Decision frameworks – when to stop, when to push
Use explicit criteria and staged investments:
- Candidate selection profile: potency, PK, safety margin, and developability must meet a Target Product Profile.
- Series-level kill rules: no clear path to exposure, non-fixable safety liability, or synthetic infeasibility.
- Portfolio thinking: balance time and capital across series. Treat each series as an option that you can exercise or abandon.
Rank chemotypes by expected LOA and cost-to-candidate, then invest in the ones that give the best information per dollar.
Common failure modes at H2L/LO
- Potent compounds with no exposure.
- Clean in vitro profile but bad metabolites in vivo.
- Chronic toxicity only apparent in longer studies.
- Solid-state or formulation problems that prevent oral dosing.
- IP or freedom-to-operate blocks after heavy investment.
The pattern repeats: teams optimize what they can measure and forget what they must measure to succeed in humans.
What comes next
Part 4 will take the candidate you pick here and explain the nonclinical studies, GLP toxicology, CMC basics, and the regulatory checklist that justify first-in-human studies.
Parts of this series
- Part 1 – From Idea to Patient: How Drug Development Actually Works
- Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- Part 3 – Hit Discovery and Lead Optimization – how teams turn a validated target into an actual drug candidate
- Part 4 – Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people
- Part 5 – Clinical Development: Phases I to III
- Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy
- Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle
Key references and sources
- Erlanson DA, et al. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016.
- High-throughput screening overview and scaling. ScienceDirect / HTS resources.
- Structure-based drug design reviews and SBDD methods. PMC review and Essays in Biochemistry.
- Pharmacokinetics and ADME basics (StatPearls, NCBI Bookshelf).
- ICH S7B and FDA guidance on nonclinical evaluation for QT risk and hERG screening.
- Jorgensen WL. Lead discovery and optimization reviews and medicinal chemistry principles.

Leave a reply to Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people – Aryan Kenia Cancel reply