
The Drug Development Playbook - Part 2
Author: Aryan Kenia
Life Sciences Analyst
This is Part 2 of The Drug Development Playbook, an seven-part series on how medicines actually move from idea to patient use and how smarter decisions at each stage change outcomes.
In Part 1, I laid out the full journey and where most programs fail. Now we start at the first real fork in the road. Before chemistry, before trials, before regulatory strategy, there’s a simpler but harder question:
What biology should we bet on?
I’ve seen strong teams lose years chasing targets that looked elegant on slides but never mattered in patients. I’ve also seen modest programs succeed because they chose the right mechanism early. This article explains how to tell the difference.
Get the companion PDF – The Consultant’s Companion: Actionable Frameworks from The Drug Development Playbook
This short guide collects the practical checklists and decision frameworks I refer to across the series. Use it in meetings, as a quick audit, or to run go/no-go gates when you don’t want to guess.
Key Summary
- Target choice determines most of a drug’s future risk and value.
- Human evidence should outweigh elegant lab data.
- One practical step this week: run a genetic reality check on every target you’re considering.
A case to consider
A biotech team once received beautiful data from an animal model. Their compound shut down a pathway, and a biomarker dropped exactly as predicted. They wanted to start preparing for Phase II.
Then they looked at human data. Patients with natural loss-of-function variants in that pathway didn’t improve clinically. Some actually did worse.
That moment changed the entire program. They pivoted to a different node in the pathway, redesigned the development plan, and avoided what would have been a very expensive failure.
That experience shaped how one thinks about target selection. If the biology doesn’t hold up in humans, nothing downstream fixes it.
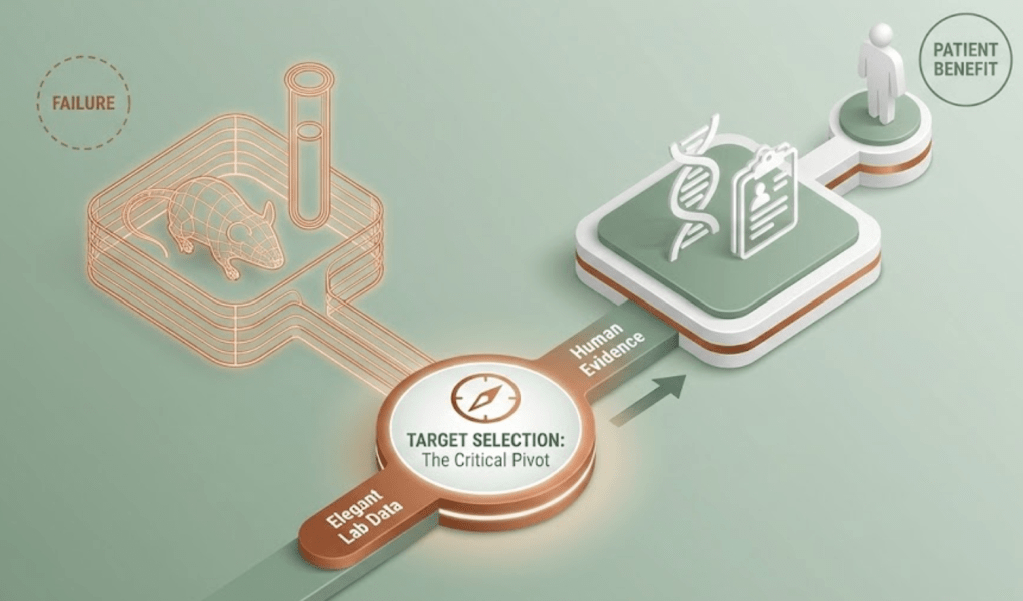
Why this decision carries so much weight
Choosing a target quietly locks in most of what follows. Screening strategy, modality, biomarkers, trial endpoints, safety assumptions, commercial positioning. Once teams commit, changing course gets harder with every month and every dollar spent.
I’ve noticed something across companies of different sizes. The strongest programs don’t rush this step. They challenge their own assumptions early, when changing direction still feels cheap.
Start with the disease, not the protein
It’s tempting to fall in love with a single gene or receptor. Real diseases rarely cooperate with that mindset.
Most complex conditions behave like networks. Different pathways drive different patient subgroups. That explains why a therapy can work brilliantly for some patients and fail in broader trials.
So the real starting point is the disease module, not the target alone.
Ask questions like:
- Which patients depend on this biology?
- What clinical change would prove we hit the right mechanism?
- How early could we see a signal that we’re on the right path?
If those answers feel vague, the science probably isn’t mature enough for a major investment.
Where credible targets actually come from
Human genetics first
Nothing beats human evidence. When nature runs an experiment for you, pay attention.
Protective mutations, disease-causing variants, and population studies reveal what truly matters in people. Some of the most successful drug classes began with these signals.
I now treat human genetics as a starting point, not a bonus.
Omics and systems biology
Modern datasets can highlight which pathways light up in disease tissue and which cell types drive pathology. They’re powerful tools for generating ideas.
But they can mislead if you treat correlation as causation. A pathway can look important simply because it reacts to disease rather than causing it.
Use these tools to ask better questions, not to declare victory.
Functional genomics
CRISPR screens and targeted perturbations let teams test whether changing a gene actually shifts disease-relevant behavior in human cells. These experiments bring you closer to causality.
Still, they work best when anchored in human observations.
Phenotypic discovery
Sometimes teams start with a phenotype and work backward to the target. This route can uncover unexpected biology. It also demands careful translation to patient relevance.
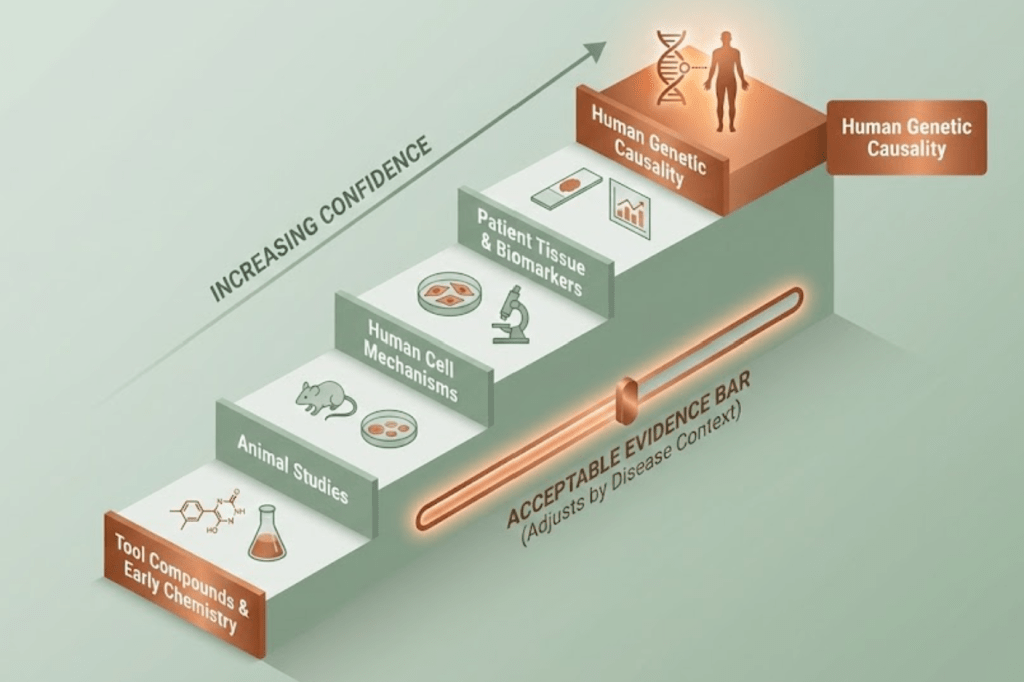
What “validated enough” looks like in practice
I like to think of evidence as a ladder:
At the top sits human genetic causality.
Below that sits data from patient tissue and biomarkers.
Then come mechanistic experiments in human cells.
Animal studies follow.
Tool compounds and early chemistry sit at the base.
The acceptable bar depends on the disease. A chronic therapy needs stronger safety confidence than a treatment for a life-threatening condition.
The key question remains simple:
If we modulate this target in humans, do we expect meaningful clinical benefit?
If the answer feels uncertain, pause and design the experiment that would clarify it.
Can we actually drug this target?
Even compelling biology can be impractical.
Teams need to check:
- Can a molecule realistically bind or modulate the target?
- Can the therapy reach the tissue where the target lives?
- Does the direction of modulation make sense biologically?
I’ve seen companies chase targets that required capabilities they didn’t have. Building new platforms mid-program rarely ends well.
Safety starts here
Target biology often predicts toxicity long before the first dose in humans.
Natural human variants again provide clues. When people live healthy lives with reduced activity of a gene, that’s reassuring. When mutations cause severe disease, caution is warranted.
Expression patterns matter too. A target active in critical organs raises the stakes.
Failure patterns that begin at target selection
- Confusing a disease marker with a driver
- Ignoring patient heterogeneity
- Trusting animal models too much
- Choosing biology incompatible with the chosen modality
- Assuming correlation equals causation
These issues surface years later as “unexpected” failures. They rarely surprise anyone who looked closely at the early data.
A practical scorecard you can use
When teams struggle to choose between targets, I suggest scoring them across five dimensions:
- Human evidence
- Mechanistic plausibility
- Druggability
- Safety plausibility
- Strategic and commercial fit
This forces clarity. It also exposes when enthusiasm outruns evidence.
One action you can take
Run a genetic reality check.
For each target on your list, ask whether human genetic evidence connects it to the disease or a related trait. If yes, it likely deserves more attention. If no, identify the fastest experiment that could clarify its role.
This single exercise often reshapes priorities.
What comes next
Part 3 moves from biology to chemistry:
Parts of this series
- Part 1 – From Idea to Patient: How Drug Development Actually Works
- Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- Part 3 – Hit Discovery and Lead Optimization – how teams turn a validated target into an actual drug candidate
- Part 4 – Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people
- Part 5 – Clinical Development: Phases I to III
- Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy
- Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle
References
Nelson MR et al., Nature Genetics 2015
King EA et al., PLOS Genetics 2019
Minikel EV et al., Nature 2024
BIO / Informa Pharma Intelligence clinical success analyses
Tufts CSDD drug development cost estimates
Cohen JC et al., NEJM 2006
Dewey FE et al., NEJM 2017

Leave a comment