
The Drug Development Playbook - Part 1
Author: Aryan Kenia
Life Sciences Analyst
Get the companion PDF – The Consultant’s Companion: Actionable Frameworks from The Drug Development Playbook
This short guide collects the practical checklists and decision frameworks I refer to across the series. Use it in meetings, as a quick audit, or to run go/no-go gates when you don’t want to guess.
Key Summary
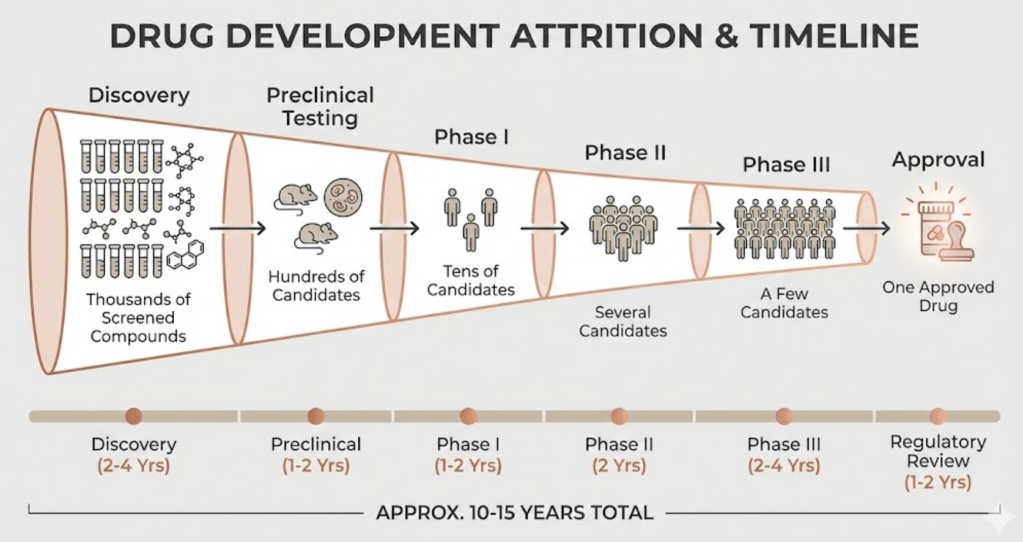
- Drug development usually takes 10 to 15 years and costs billions; only about 1 in 10 programs entering human testing reach approval.
- Most value is set or lost before late-stage trials, with Phase II causing the largest drop in programs.
- This series shows the step-by-step playbook I use as a life science consultant to sequence decisions, reduce avoidable risk, and preserve value.
This is a series. Here’s what it is and why it matters
You’re reading the first article in The Drug Development Playbook, a seven-part series I wrote to help scientists, R&D leads, investors, and consultants make smarter decisions across the whole journey from idea to patient. One article teaches the phases. A series teaches the moves that actually change outcomes.
Each part focuses on a single stage and gives concrete tools you can use immediately. If you want to jump ahead, pick a part below and follow the link.
- Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- Part 3 – Hit Discovery and Lead Optimization – how teams turn a validated target into an actual drug candidate
- Part 4 – Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people
- Part 5 – Clinical Development: Phases I to III
- Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy
- Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle
I’ll link back to these parts naturally as we move through the map, so readers can hop to the playbook that fits their next decision.
A short example
A mid-stage biotech ran a tidy Phase I and believed the path to Phase III was clear. The team planned a large Phase II with a broad patient population, a single unvalidated endpoint, and a dose chosen from one small PK run. An external consultant reviewed the plan and flagged three hard problems that would likely waste money and time: the endpoint would miss the expected biology, the population would dilute any signal, and the dose had no real dose-response data behind it. The company paused, redesigned the Phase II to include an early biomarker cohort and an adaptive dose arm, and moved forward with much clearer decision gates. That is exactly the kind of fix this series will teach you to make.
The headline numbers to keep in your head
These figures explain why teams behave the way they do and where consultants add value.
- Typical time to market: 10 to 15 years.
- Probability of approval from Phase I: roughly 8 to 12 percent.
- Industry estimates of full development cost per approved drug: commonly in the multi-billion USD range.
For the regulatory framework, see the U.S. regulator for the staged view of development and review. U.S. Food and Drug Administration
For lifecycle and centralized procedures in Europe, see the EU regulator. European Medicines Agency
ICH guidance frames study design quality as a core principle. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
Why drug development feels impossible
Biology fights complexity
A target can behave perfectly in a cell line or mouse and still fail in a heterogeneous human population. Pathway compensation, disease subtypes, and chronic disease dynamics all change how an intervention performs. You need multiple, staged experiments to learn enough to act.
Regulation demands the right evidence, in the right order
Regulators ask for evidence that supports safety and benefit in real patients. That expectation drives endpoint choice, sample size, and manufacturing controls. Design early or pay later in time and money.
Capital multiplies mistakes
You spend small sums early to buy information. You spend large sums later to confirm it. If you scale before you have clear proof of concept, you multiply financial risk. Most of an asset’s value depends on decisions you make when uncertainty runs highest.
A short map of the series
I’ll cover each of these in depth. I add the link to the part you’ll want when you face that decision.
Part 1 – From Idea to Patient: How Drug Development Actually Works
- What it is: a compact, consultant-minded overview of the whole lifecycle, the hard numbers, and where value gets created or lost.
- Use it when you need the big-picture decision map.
- Read the playbook: [Part 1 – From Idea to Patient]
Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- What it is: a hands-on target evaluation playbook – how to test causal biology, prioritize targets, and set kill criteria.
- Deliverable you can use: target validation checklist and short decision tree.
- Read the playbook: [Part 2 – Target Identification and Validation]
Part 3 – Hit Discovery and Lead Optimization
- What it is: practical guidance on screening, hit triage, SAR cycles, early ADME filters, and when to stop a chemistry series.
- Deliverable you can use: hit-to-lead gating checklist and a template for early DMPK go/no-go.
- Read the playbook: [Part 3 – Hit Discovery and Lead Optimization]
Part 4 – Preclinical Development and IND/CTA Readiness
- What it is: the IND/CTA enabling package playbook – GLP tox, PD/PK, biomarker selection, and CMC basics you must have.
- Deliverable you can use: IND readiness checklist and a mock pre-IND briefing slide deck.
- Read the playbook: [Part 4 – Preclinical Development and IND/CTA Readiness]
Part 5 – Clinical Development: Phases I to III
- What it is: how to design Phase I for informative PK/PD, run Phase II as a true proof-of-concept, and set up Phase III for regulatory and payer success.
- Deliverable you can use: adaptive Phase II template, dose-finding subprotocol, and primary endpoint decision flow.
- Read the playbook: [Part 5 – Clinical Development: Phases I to III]
Part 6 – Regulatory Submission and Review (NDA/BLA/MAA)
- What it is: a step-by-step guide to filing the dossier, running pre-sub meetings, and using accelerated pathways appropriately.
- Deliverable you can use: CTD-module checklist and a regulator meeting memo template referencing ICH and agency expectations.
- Read the playbook: [Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy]
Part 7 – Post-marketing, Lifecycle Management, Generics and Biosimilars
- What it is: practical steps for pharmacovigilance, label lifecycle plays, and the specific routes for generics and biosimilar entrants.
- Deliverable you can use: post-marketing study plan template and an RWE playbook for label expansion.
- Read the playbook: [Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle]
Where programs actually fail, and what I tell teams to do about it
Phase II causes the most value destruction. The usual failure modes repeat.
Common problems
- Wrong endpoint that misses the biology.
- Heterogeneous patient mix that washes out effect.
- Dose chosen from incomplete PK/PD data.
- CMC surprises that force delayed launches.
What I make teams commit to before Phase III
- A robust dose-response from Phase II.
- A primary endpoint that maps to mechanism and regulator expectations.
- A CMC bridge proving commercial material equals clinical material.
- Clear stop/go criteria so the team will stop fast if thresholds fail.
Discipline here saves tens or hundreds of millions.
How I read the funnel differently than a scientist
Scientists focus on mechanism. I focus on the decision you face next.
- I rank assets by risk-adjusted NPV and by the capacity required to run the next stage.
- I force the team to state the single decision the next study must answer.
- I align clinical, regulatory, and CMC teams early so the dossier reads as one coherent story and not like three separate projects.
If you want to think like a consultant, ask: what is the single question we must answer next, and what is the cheapest, fastest study that answers it?
A reusable checklist you can use now
- Do we have a clear objective for the next study?
- Have we predefined stop/go criteria and budget thresholds?
- Does the inclusion criteria increase signal or noise?
- Do we have a CMC plan that can scale to commercial manufacturing?
- Have we sought regulatory advice on endpoints and patient population?
What’s next and how the series flows forward
Part 2 will go deep on target identification and validation and include a validation checklist you can run in-house. From there I’ll move through hit discovery, preclinical readiness, trial design, regulatory filings, manufacturing scale-up, and lifecycle strategy. You’ll get a mix of checklists, visual templates, and short case studies you can reuse with clients or include in your portfolio.
Parts of this series
- Part 1 – From Idea to Patient: How Drug Development Actually Works
- Part 2 – Disease Understanding and Target Identification: Picking the Biology Worth Backing
- Part 3 – Hit Discovery and Lead Optimization – how teams turn a validated target into an actual drug candidate
- Part 4 – Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people
- Part 5 – Clinical Development: Phases I to III
- Part 6 – From Pivotal Data to Approval: The Regulatory Review Strategy
- Part 7 – After Approval: Running Safety, Real World Evidence, and the Product Lifecycle

Leave a reply to Preclinical Development and IND/CTA Readiness: making the candidate safe enough to test in people – Aryan Kenia Cancel reply